Ocular melanoma
More than 95% of all ocular melanomas occur intraocularly, the remainder arising in conjunctiva. These two sub-types of ocular melanoma are completely different from each other in their molecular biology and natural history.
Overview
Intraocular melanomas arise from melanocytes in the uvea. The uvea is a vascular, pigmented tissue comprising: (A) the cup-shaped choroid, which lies between the retina and the sclera; (B) the ciliary body, a muscular ring of tissue, which lines the rim of the choroid, adjusting the lens focus; and (C) the iris, which lies anterior to the lens, altering the pupil size. About 90% of uveal melanomas involve choroid, with about 5% arising in the ciliary body and 5% developing in the iris.
Epidemiology
Uveal melanomas account for about 5-10% of all melanomas. They rarely present before adulthood, the incidence increasing with age to peak at around sixty years [1]. There is no sex preponderance. Risk factors include: fair skin, blue or grey iris color, increased number of cutaneous naevi, congenital ocular melanocytosis, and neurofibromatosis. The role of sunlight is disputed.
Clinical features
Choroidal and ciliary body melanomas cause exudative retinal detachment, which results in blurred or distorted vision, visual field loss and flashing lights. In addition, ciliary body melanomas press on the lens to cause astigmatism and cataract. Iris melanomas usually form a visible nodule. All type of uveal melanoma can show diffuse growth, which can cause glaucoma by obstructing aqueous fluid outflow through the drainage meshwork in the anterior chamber angle. Uveal melanomas can spread extraocularly along small blood vessels and nerves exiting the sclera. Optic nerve invasion is rare. Advanced melanomas can make the eye blind and painful as a result of retinal detachment, glaucoma and inflammation. About 30-40% of all uveal melanomas are asymptomatic at the time of detection, being discovered as a result of routine ocular examination.
Pathology
The two uveal melanoma cell types are spindle and epithelioid, with most tumors being of mixed cell type. Lymphocytes and macrophages are often present, although the numbers vary greatly from tumor to tumor. Periodic-acid-Schiff staining shows a variety of interstitial tissue patterns, such as so-called “closed loops” [2]. The microvascular density is variable [3]. In Caucasians, most uveal melanomas are amelanotic or lightly pigmented, with choroidal and ciliary body melanomas appearing dark brown or grey only because of the overlying pigment epithelium. Uveal melanomas tend to show chromosomal abnormalities such as partial or total loss of chromosome 3 (i.e., monosomy 3), gains in chromosome 8 (e.g., trisomy or isochromosome 8q), and gains in chromosome 6 (e.g., isochromosome 6p) [4, 5]. Uveal melanomas metastasize via the blood stream (i.e., hematogenously); lymphatic spread to regional nodes is very rare, occurring only if the tumor has spread extraocularly to involve conjunctiva [6]. Metastatic disease involves the liver in more than 90% of patients, other relatively common sites being skin, lung, and bone. Most patients with metastases die within a year of the onset of symptoms [6].
Prognostic factors
The most accurate predictor of metastatic disease in uveal melanoma is detection of monosomy 3 [7]. Almost all tumors with this cytogenetic abnormality are fatal as compared to less than 5% of tumors without chromosome 3 deletion. Chromosome 8 gains are associated with monosomy 3 and apparently occur at a later stage in tumor development, so that their presence indicates a worse prognosis. Chromosome 6p gains rarely coincide with monosomy 3 and, therefore, indicate a good survival probability [5]. Monosomy 3 is believed to develop at a very early stage in tumor growth [8]; however, tumor heterogeneity can occur, with implications for biopsy [9]. Epithelioid cell type indicates a poor prognosis, firstly because it correlates strongly with monosomy 3 and perhaps because in the presence of monosomy 3 it indicates more rapid growth. Other histological factors indicating an increased risk of metastasis include: closed loops; lymphocytic infiltrates; macrophages; and high microvascular density. Basal tumor diameter correlates with poor survival, firstly, because large tumours are more likely to show monosomy 3 and, secondly, because of lead-time bias. Tumor thickness is not significant in multivariate analyses Ciliary body involvement correlates with monosomy 3 and with poor survival, although these associations are unexplained. Extraocular spread is associated with increased mortality, mostly because it indicates that the underlying intraocular tumor is more malignant.
Staging
The present TNM (Tumor, Node and Metastasis) staging system is based on basal tumor diameter, tumor thickness, ciliary body involvement and extraocular spread [10]. It is currently being revised. It is not widely used by ocular oncologists. An alternative approach is being evaluated, which uses a neural network to: (A) take account of normal life-expectancy as determined by age and sex; (B) determines whether or not the tumor has metastatic potential; (C) stages lethal tumors to deal with lead-time bias; and (D) adjusts for tumor growth rate.
Ocular therapy
For many years, it was believed that ocular treatment was life-saving, especially with large uveal melanomas, which were, therefore, treated more radically, by enucleation. Eye-conserving treatments such as radiotherapy were considered risky. In 1978, Zimmerman hypothesized that enucleation accelerated metastatic death and this suggestion prompted measures such as pre-enucleation radiotherapy [11]. In the USA, the Collaborative Ocular Melanoma Study (COMS) reported that it found no difference in survival: (A) between pre-enucleation radiotherapy and enucleation alone; and (B) between brachytherapy and enucleation [12, 13]. A recent re-appraisal indicated that the COMS was essentially inconclusive, because of insufficient data [14]. Small melanomas were considered to be relatively benign and were often left untreated for months or years until growth was observed [15]. There is growing circumstantial evidence that monosomy 3 uveal melanomas start to metastasize at a very early stage, perhaps even when they are indistinguishable from naevi [16]. The implication of this view is that ocular treatment is only palliative, except perhaps with very small tumors. The problem is that only about 25% of small melanomas have metastatic potential. Several centers have recently started performing biopsy on small melanomas, to determine whether or not monosomy 3 is present; however, biopsy does not always produce an adequate sample and sometimes causes vitreous hemorrhage and other complications. Fluorescence in situ hybridization (FISH) is most widely used but being replaced by multiplex ligation probe amplification (MLPA) and other methods that provide more information from smaller samples. In most centers, the first choice of therapy for uveal melanomas is brachytherapy, delivered using an iodine or ruthenium plaque [17]. Proton beam is available in only a few centers, where it is used either in selected cases, when brachytherapy is not possible, or for all patients (e.g. Lausanne, Boston). A few centers advocate stereotactic radiotherapy. There are appreciable ocular complications after radiotherapy, with loss of vision and the eye itself. Such morbidity has been attributed to collateral damage to healthy ocular tissues. Recent evidence indicates that in many patients, the ocular morbidity occurs because the irradiated tumor can become toxic to the eye, as a result of exudation and the release of angiogenic factors [18]. These are treatable with methods such as: intraocular injections of steroids and/or anti-angiogenic agents; trans-pupillary thermotherapy or photodynamic therapy of the melanoma or local excision of the tumor, either trans-retinally or trans-sclerally. Primary enucleation now tends to be performed only if the tumor involves optic nerve or if it is very large, exceeding 17 mm in basal diameter. In practice, ocular conservation is attempted in about 70% of patients and successful in about 90% of these [19].
Screening for metastases
Systemic screening is performed mostly in the hope of detecting resectable hepatic metastases [6]. Methods include liver imaging, such as ultrasonography, and biochemical liver function tests, which are performed every six or twelve months, indefinitely [20]. Chest radiography is not useful, except when metastasis has been detected. Positron emission tomography (PET) is useful for determining the extent of systemic disease, for example, if partial hepatectomy is being considered [21]. There is no consensus as to whether screening should be performed in all patients or only in those with increased risk.
Systemic therapy
There is a growing desire for adjuvant systemic therapy in high risk uveal melanoma patients; however, no randomized, prospective studies have yet shown any benefit. Obstacles to such studies include the rarity of uveal melanoma and the limited number of ocular oncology centers, which perform cytogenetic studies on a routine basis, required to distinguish between lethal and non-lethal tumors. Long term survival has been reported after apparently complete resection of hepatic metastases. Encouraging results have also occurred after intra-hepatic chemotherapy. Systemic chemotherapy has generally been disappointing [6].
CONJUNCTIVAL MELANOMA
Overview
Most conjunctival melanomas arise in the bulbar (or ‘ocular’) conjunctiva, usually at the limbus (i.e., edge of the cornea) and most often temporally [22]. A minority develop in the caruncle (i.e. fleshy pink tissue at the nasal end of the conjunctiva) or in the palpebral conjunctiva (i.e., inner surface of eyelid). Knowledge on conjunctival melanomas is diminished by the fact that most patients are referred to an ocular oncology centre only after initial treatment by general ophthalmologists, who may not perform adequate treatment and who often provide incomplete baseline data.
Epidemiology
Conjunctival melanomas account for less than 5-10% of all ocular melanomas. They usual present in adulthood [22]. There is no sex preponderance. The most important risk factor for invasive melanoma is, perhaps confusingly, referred to as ‘primary acquired melanosis (PAM) with atypia’. This is essentially ‘conjunctival melanocytic dysplasia’, culminating in ‘in situ melanoma’ and, ultimately, invasive melanoma [23]. Some melanomas are believed to arise from naevi [23].
Clinical features
Conjunctival melanomas can be nodular or diffuse, that is, thick with discrete margins or thin and wide with indistinct edges. Many are mixed (i.e., nodulo-diffuse). There may be extensive intra-epithelial disease, consisting either of the pre-existing melanocytic dysplasia, or secondary pagetoid spread from the melanoma. The tumor color can be black, grey, brown, tan, pink or white. A minority of tumors are multinodular. Spread can occur to adjacent eyelid skin, naso-lacrimal duct, orbit, eye, nasal sinuses and intracranial cavity. Metastatic spread can occur to regional lymph nodes and systemically. There is circumstantial evidence that seeding can occur iatrogenically, if tumor biopsy or excision is not performed properly [24].
Pathology
Primary acquired melanosis (PAM) requires histology with immunohistochemistry to distinguish: (A) racial melanosis (i.e., increased melanin production from melanocytes that are normal in shape, location and number); (B) ‘PAM without atypia’, which consists of melanocytic hyperplasia, with the melanocytes showing an increased density, without atypia or displacement from the basal layer; and (C) ‘PAM with atypia’, consisting of melanocytic dysplasia, i.e., melanocytes with cellular atypia infiltrating with increasing density, partial or the entire conjunctival epithelial thickness. Invasive melanoma shows spread into the lamina propria of the conjunctiva. Invasive melanoma can be associated with diffuse infiltration either within the conjunctival epithelium, subepithelially and into adjacent tissues.
Prognostic factors
The overall ten-year mortality is reported to be 25-30% [25-27]. Risk factors for metastatic spread are: involvement of non-bulbar conjunctiva, especially caruncle; local tumor recurrence; multi-focal tumors; increased tumor thickness; high mitotic rate; epithelioid tumor cell type; and lymphatic invasion.
Staging
The present TNM staging system categorizes conjunctival melanomas as: T1, bulbar tumors; T2, bulbar tumors involving cornea; T3, involvement of non-bulbar conjunctiva; and T4, involvement of eyelid skin, eye, orbit, nasal sinuses or brain. This system has several limitations. First, there is no evidence that corneal involvement is associated with a worse prognosis for survival. Second, it ignores basal tumor diameter, which is important. Third, it takes no account of quadrant, whereas nasal tumors have a worse prognosis than temporal disease. Fourth, it assumes that any non-bulbar conjunctival disease indicates a poor prognosis, whereas caruncular involvement is particularly serious. Finally, in T4, it stages all routes of spread equally, whereas intracranial disease is likely to have a worse prognosis than eyelid involvement. The TNM system is currently being revised and should correct most of these weaknesses in the new version.
Ocular therapy
Nodular tumors are treated by local excision with adjunctive brachytherapy or cryotherapy. If unresectable, such tumors can be treated with brachytherapy or some form of external beam radiotherapy. Intra-epithelial disease is treated with mitomycin C drops or some other form of topical chemotherapy. Exenteration is reserved for uncontrollable disease.
Screening for metastases
The role of sentinel lymph node biopsy is controversial [28] Not all patients with systemic metastases have clinical evidence of regional lymph node involvement. Other systemic screening is the same as for cutaneous melanoma.
Systemic therapy
Systemic therapy for metastatic disease from conjunctival melanomas is the same as for metastases from cutaneous primaries.
CLINICAL PRACTICE GUIDELINES
TABLE 1: Summary of evidence and recommendations for choroidal and conjunctival precursor melanocytic lesions (*).

TABLE 2-A: Summary of evidence for choroidal melanoma (*).

TABLE 2-B: Summary of recommendations for choroidal melanoma (*).

TABLE 3: Summary of evidence and recommendations for conjunctival melanoma (*).

TABLE 4: Summary of evidence and recommendations for eyelid melanoma, orbital melanoma and psychological aspects of ocular melanoma (*).

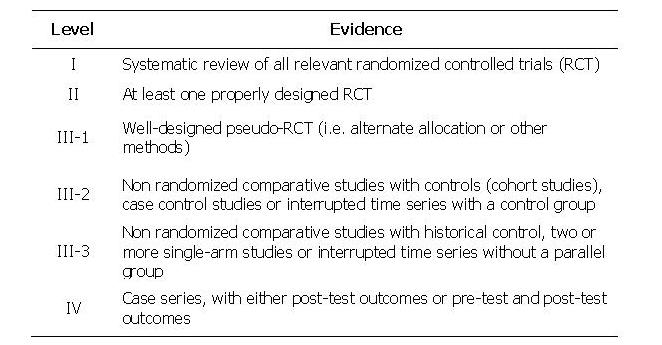
TABLE 5: Levels of evidence used in the above Tables. Grades of recommendations (A: excellent, B: good, C: satisfactory, D: poor) are based on level of evidence, consistency across studies, clinical impact and applicability.
TNM STAGING SYSTEM FOR OCULAR MELANOMA
FIGURE 1: AJCC TNM staging of conjunctival melanoma

FIGURE 2: AJCC TNM staging of uveal melanoma (choroid)

FIGURE 3: AJCC TNM staging of uveal melanoma (iris and ciliary body)
