Risk factors
[1] Sun exposure
[2] Dysplastic nevi
[3] Age
[4] Race
[5] Personal or family history of melanoma
[6] Phototype
[7] Xeroderma pigmentosum
[8] Li-Fraumeni syndrome
[9] Hereditary retinoblastoma
[10] Immune suppression

Sun exposure
The risk of melanoma increases with intermittent intense sun exposure but the relationship between melanoma and sun exposure is very complex in view of host factors such as skin and hair color [1]. By contrast, chronic sun exposure - which is well known to correlate with the risk of epithelial skin cancers - may even protect from developing melanoma [2, 3]. Animal studies and population-based studies point to the ultraviolet(UV)-B radiation emitted by the sun as a major exogenous causative factor but again whether UVB is more harmful than UVA is still not really established [4-6]. Evidence also exists that melanoma patients are greater users of artificial UV sources than the general population, although no general consensus exists as of the few studies which found a positive relationship the magnitude of the relative risks was small[7, 8]. However, the correlation between UV exposure and melanoma incidence is not completely clarified, as suggested by the fact that melanoma does not show a preference for skin parts exposed to sunlight, unlike epithelial skin malignancies. Moreover, although melanin is believed to protect human skin against the mutagenic effects of UV radiation [9], the development of melanoma among albinos is a very rare occurrence [10], which further questions the role of UV light in melanoma genesis. Finally, as regards the relationship between the use of sunscreens and the risk of melanoma, the results of epidemiological studies do not support their protective effect so far but this may take a long time to be properly assessed in view of the long lag time for melanoma [11, 12]. A multifactorial model of cancerogenesis with complex interactions between genes and environment is likely to be the explanation for these apparently conflicting evidences.
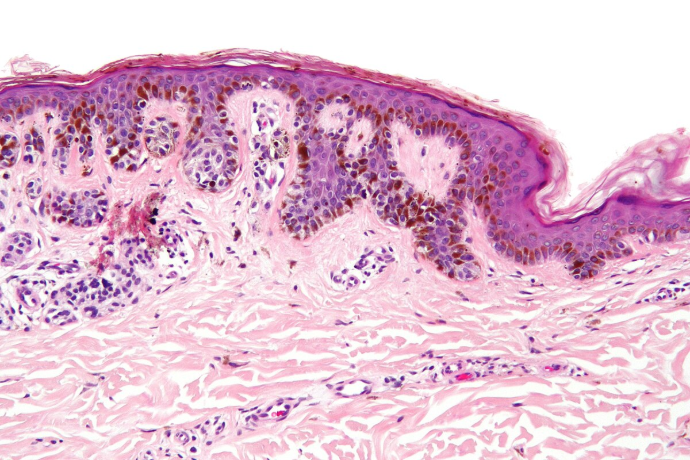
Dysplastic nevi
Dysplastic nevi are characterized histologically by the presence of an architectural disorder with some fusion of rete ridges, lymphocytic infiltrate in the upper dermis and nuclear atypia of the melanocytes in the nest [13, 14]. However dysplastic naevi are also often recognisable clinically and usually have a size of equal or above 5 mm in diameter with a hazy border and irregular pigmentation (clinically defined as atypical naevi). Individuals with atypical naevi are at a higher risk of melanoma [15], which does not necessarily arise in the atypical lesion but can also appear on normal skin. Therefore, atypical nevi can be considered markers that allow the identification of individuals at an increased risk for melanoma. Naevi do appear with age with a steady increase in childhood and early adulthood and thereafter a steady decline from middle age onwards. This senescence of naevi with age is interesting and the role of senescence genes such as p16 is suspected to play a role in the disappearance of naevi with age [16]. Naevi number has also been correlated with telomere length with higher number of naevi in subjects with longer telomers suggesting that naevi may also be a good marker of ageing [17]. There has been some advances in looking for genetic factors in the causation of naevi. Variation in naevi number is in most part explained by genetic factors and twin studies have shown that 60% of this variance is due to additive genetic factors. Recent collaborations are leading to genome wide association studies looking for common low penetrance genes influencing naevi numbers in Caucasians and interesting candidates are currently being investigated. Naevi number is a very useful intermediate phenotype for melanoma as it is to date the highest risk factor for this tumor and this trait can easily be documented in all Caucasian populations whilst melanoma is rare. There are, however, some genome wide association studies now being performed looking for common polymorphism conferring an increased risk of melanoma and these studies have gathered blood DNA from melanoma cases from all over the world.
Age
Melanoma commonly afflicts young, productive members of the society, with one-fourth of melanoma cases diagnosed in the United States occurring in individuals before the age of 40. Melanomas diagnosed in those below 40 years of age are more likely to be thinner tumors with a better prognosis. However there is an increase of melanoma with age like all other cancers. Although all ages are at risk, melanoma is extremely rare prior to puberty; thereafter the incidence increases with age, the median age at diagnosis ranging from 45 to 55 years [18].
- Race
Melanoma is much more frequent in whites, the proportion between Caucasians and Asian/Black populations being 20 : 1 [18].
In non-whites melanoma is rare and mostly confined to non pigmented sites such as the subungual regions, the palms of the hand and the soles of the feet. Although the incidence is lower, the mortality rate in non-white patients is higher, which might be due to late diagnosis.
- Personal or family history of melanoma
Patients with melanoma have an increased risk of developing a second melanoma. The incidence of multiple primary melanomas ranges from 1.3% to 8.0% in large retrospective series [19].
People with first-degree relatives affected with melanoma are at higher risk for melanoma [20] (see also the "Familial melanoma" section).
- Phototype
People with fair complexion, blue gray or green eyes, red or blond hair, and lots of freckles are at higher risk of developing melanoma as compared to people with other skin types which is approximately a two fold increase in individuals with red and blond hair compared to those with dark hair [20]. Since this risk correlates with that of sensitivity to sunlight, investigators have devised a phototype classification that takes into account these features [21], as shown in Table 1.
Xeroderma pigmentosum (XP) is a rare genetic disease inherited in an autosomal recessive manner (its estimated prevalence is 1 : 1,000,000 in the United States and 1 : 100,000 in Japan).
XP is characterized by sun sensitivity, ocular damage and a 1,000-fold increased risk of cutaneous (basal cell carcinoma, squamous carcinoma as well as melanoma) and ocular neoplasms [22].
XP is known to be associated with mutations in the following genes: XPA, ERCC3 (XPB), XPC, ERCC2 (XPD), DDB2 (XPE), ERCC4 (XPF), ERCC5 (XPG), and POLH (XP-V). Mutations in XPA and XPC account for about 50% of XP cases. These genetic alterations are responsible for impaired nucleotide excision repair, which ultimately leads to genomic instability and carcinogenic mutations following DNA damage by ultraviolet radiation.
Currently no cure exists for XP. The DNA damage is cumulative and irreversible. Management is limited to avoidance of exposure to damaging ultraviolet light by staying indoors with sunlight blocked out, and by means of protective clothing, sunscreens and sunglasses.
TP53 encodes p53, the so called "genome guardian" because of its pivotal role in cell cycle arrest following DNA damage, which preserves cells from propagating errors in the genetic code. Somatic mutations of this gene can be found in about 50% of all sporadic tumors. Furthermore, germline mutations of TP53 can be found in the Li-Fraumeni syndrome (LFS), a cancer syndrome characterized by the predisposition to a wide range of tumors types [23]. Classical LFS is defined by: 1) a proband with sarcoma diagnosed before 45 years; 2) a first degree relative aged under 45 years with any cancer; 3) an additional first- or second-degree relative in the same familial line with any cancer aged under 45 years or a sarcoma at any age. Mutations in TP53 are found in 77% of classical LFS families. Many studies have indicated an association with a wider range of tumors, including melanoma [24-26], but since the absolute number of melanoma cases is low, there is some debate regarding whether this tumor type is truly a rare manifestation of LFS. There is also some evidence that subjects belonging to other family cancer syndromes with an excess of all cancers in general may be more prone to melanoma and this is particularly relevant for families with pancreas, brain and breast cancers. Other family cancer syndromes such as Neurofibromatosis have also been associated with a higher prevalence of melanoma.
The RB1 gene was the first tumor suppressor gene to be cloned [27]. Knudson's classical two-hit hypothesis is based on the observation that individuals carrying RB1 germline mutations develop retinoblastoma when the remaining wild-type copy is lost somatically. The penetrance of this cancer susceptibility gene germline mutations is approximately 90%, and tumors are generally bilateral. Hereditary retinoblastoma occurs in the early childhood and the 5-year survival rate is about 90% if the tumor is diagnosed when still confined to the eye. The activity of the retinoblastoma protein (pRb) - a well characterized cell cycle inhibitor - is regulated by p16INK4A through its negative effect on the activity of the cyclin D-CDK4/CDK6 complexes, which phosphorylates and thereby inactivates the transcriptional suppressor function of pRb. An excess risk of cancers other than retinoblastoma has been reported in RB1 germline mutation carriers, with a cumulative incidence of adult cancer of approximately 70% [28]. Besides an increased risk of epithelial cancers in older adults, this figure is largely due to the early onset sarcomas and melanoma: in particular, melanoma accounts for about 7-8% of non-retinoblastoma malignancies in this cancer-prone population [28-30].
Like other cancer types, there is evidence that melanoma development can be favored by immune suppression [31]. This is particularly evident in transplant recipients chronically receiving immunosuppressive agents but whilst the risk of squamous cell carcinoma is 100 fold for melanoma the risk is much smaller with a relative risk of 1.5 to 2 [32-34].